http://dx.doi.org/10.18273/revsal.v52n1-2020007
Reporte
de caso
Translocación cromosómica no
balanceada t(5;7)(q22;p15) en un
niño con anomalías congénitas:
reporte de caso clínico
Unbalanced chromosomal translocation
t(5;7)(q22;p15) in a child with congenital anomalies: clinical case report
María Amparo Acosta Aragón1
Julián Andrés Hamdan Pérez1
Luisa María Morán Quiñones1
Diana Catherine Moreno Ortega1
1. Universidad
del Cauca. Popayán, Colombia.
Correspondencia: Julián Andrés Hamdan. Dirección:
calle 5ª 21-94 B/José María Obando. Popayán. Colombia. Teléfono: +57 310
6362325.
Correo electrónico: julianhp101@gmail.com
Resumen
Introducción:
La incidencia de las anomalías congénitas es de 0,5% dentro de los cuales el
0,1-0,3% corresponden a anomalías cromosómicas estructurales, entre ellas están
las translocaciones no balanceadas en las que hay pérdida o ganancia de
información genética que da como resultado manifestaciones fenotípicas con
compromiso en la salud de quienes las padecen.
Reporte de
caso: Se describe un paciente escolar con una translocación no balanceada
t(5;7) (q22;p15) de origen paterno y sus repercusiones.
Discusión:
Cuando existen reordenamientos en el material genético, las manifestaciones
clínicas están ligadas a la localización de los puntos de ruptura y como
consecuencia a los genes que estén incluidos en estos segmentos, tal como se
presentó en nuestro caso índice. Conclusiones: Es importante el estudio de
estos pacientes ya que deben permanecer en vigilancia médica por el riesgo de
desarrollar patologías relacionadas con alteraciones en los genes implicados en
el reordenamiento genético.
Palabras
clave: Translocación; anomalías congénitas; malformaciones congénitas;
cariotipo; cromosoma 5; cromosoma 7.
Abstract
Introduction: The incidence of congenital anomalies
is 0,5%, wich 0,1 to 0,3% belong to structural chromosomic anomalies, between
these are unbalanced translocations in which there are loss or gain of genetic
information that results in phenotypic manifestations with health compromise of
whom suffer it.
Case report: A scholar patient with an unbalanced
translocation t(5;7) (q22;p15) of paternal origin and its repercussions is
described.
Discussion: When there are rearrangements in genetic
material, the clinical manifestations are linked to breakpoints localizations
and as consequence to the genes included in this segments, as presented in our
index case.
Conclusions: The study of these patients is important
because they must remain under medical surveillance due the risk of developing
pathologies related with gene alterations implicated in the genetic
rearrangement.
Keywords: Translocation; congenital anomalies;
congenital malformations; karyotype; chromosome 5; chromosome 7.
Recibido: 07/06/2019
Aprobado: 18/11/2019
Publicado online: 12/12/2020
Introducción
Las anomalías congénitas o anomalías cromosómicas
estructurales (translocaciones, deleciones, inversiones, duplicaciones,
isocromosomas, cromosomas en anillo, entre otras)1,2, determinan cambios en el genoma
originando los denominados síndromes cromosómicos o cromosomopatías causantes
de diversos problemas en la salud. La presentación clínica dependerá de los
cromosomas y los segmentos implicados en dicha reorganización genómica3.
La Universidad de Quebec (Canadá) determinó que la
incidencia de las aberraciones cromosómicas es de 0,5% dentro de los cuales el
0,1 a 0,3% corresponden a anomalías cromosómicas estructurales4; la incidencia de estas
últimas es de 1 por cada 1000 diagnósticos prenatales – University of Melbourne
and Oxford University5.
Las translocaciones balanceadas de los autosomas ocurren en
1 de cada 500 recién nacidos6,
siendo éstas el reordenamiento estructural más común de los seres humanos4; gran parte de las
translocaciones son heredadas y en un 0,02% tienen un origen de novo7.
Estas translocaciones son causa de anomalías congénitas, las
cuales tienen una frecuencia del 0.7 al 1.5% en los recién nacidos8 y causan la muerte de
hasta 270.000 lactantes menores, además de ser responsables de aproximadamente
3,2 millones de discapacidades anuales2. Muchas de estas alteraciones se detectan
durante los controles prenatales o mediante la realización de un cariotipo a
los progenitores de pacientes anormales que tienen translocaciones no
balanceadas5.
Una translocación es balanceada cuando en el intercambio de
material genético no hay pérdida ni ganancia de este, el individuo es
aparentemente normal, aunque hasta en un 5% de los portadores se pueden
presentar anomalías fenotípicas con o sin alteraciones mentales o problemas de
fertilidad atribuídas a anomalías submicroscópicas de tipo deleción,
duplicación o disrupciones genómicas5.
Por otro lado, en los individuos con translocaciones no
balanceadas, en el intercambio cromosómico se presenta pérdida o ganancia de
información genética lo que da como resultado manifestaciones fenotípicas con
algún grado de retraso mental, retraso en el desarrollo, del aprendizaje,
subfertilidad o malformaciones
congénitas6,9.
El presente texto tiene por objetivo describir un caso
clínico de manera detallada, el cual hace referencia a un escolar que presenta
una anomalía cromosómica estructural de tipo translocación no balanceada t(5;7)
(q22;p15) de origen paterno, además de mencionar las repercusiones funcionales
y estructurales que tuvo dicha cromosomopatía en el paciente y una discusión de
la literatura sobre el tema. Para presentar este reporte de caso se solicitó la
autorización y el consentimiento informado de los padres del menor de edad
estudiado.
Reporte del caso
Escolar de 9 años y 6 meses de edad, sexo masculino,
procedente del área rural del Tambo, Cauca (Colombia), hijo de madre de 37 años
con fórmula obstétrica G2C1A1 y padre de 41 años de edad quienes negaron tener
cualquier tipo de consanguinidad. El primer embarazo de la misma unión terminó
en aborto espontáneo a las siete semanas de gestación y su segundo embarazo se
finalizó por cesárea a las 38 semanas indicada por preeclampsia severa. Su
peso al nacer fue de 2900 gramos y la talla de 49 cm, sin requerimiento de
hospitalización perinatal.
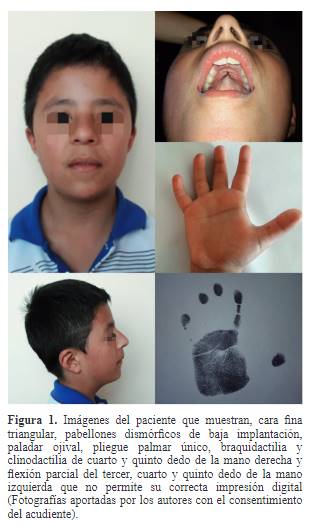
Dentro de los antecedentes de importancia destacan hallazgos
clínicos detectados a los 23 meses de edad como lo son: retraso en el
neurodesarrollo con dismorfismo facial, estrabismo divergente, mano derecha con
pliegue palmar único, braquidactilia, clinodactilia bilateral del cuarto y
quinto dedo e hipotiroidismo subclínico, este último ha requerido manejo con
Levotiroxina 25 mcg/día y control con endocrinología pediátrica hasta la
actualidad. A los cinco años de edad se le diagnosticó un índice general
cognitivo muy bajo (Coeficiente intelectual – CI: 52),

Tras la valoración clínica inicial, y en conjunto con las
múltiples valoraciones realizadas por un grupo interdisciplinario de
profesionales que se llevaron a cabo en un tiempo promedio de 8 años (las
remisiones y estudios más relevantes se resumen en la Tabla 2), el
diagnóstico presuntivo del paciente fue un síndrome cromosómico de tipo
numérico o estructural, sustentado por sus características clínicas, motivo por
el cual se hizo uso de la citogenética que estudia, diagnostica y esclarece las
enfermedades de herencia cromosómica así como innumerables síndromes
dismórficos, facilitando no solo el asesoramiento genético del paciente y sus
familias sino también el pronóstico de ellas; dentro de sus indicaciones está
la presencia de dismorfismos, retardo mental de origen desconocido y la
confirmación diagnóstica de enfermedades de herencia cromosómica numéricas o
estructurales, razón por la cual se le realizó un cariotipo con bandeo Q y G al
paciente en estudio10,
diagnosticando una anomalía cromosómica estructural tipo translocación no
balanceada entre los cromosomas 5;7 (q22;p15) de origen paterno causante de
las anomalías congénitas tanto de tipo funcional evidenciado por su
hipotiroidismo congénito, su discapacidad mental y trastorno de la conducta
como TDAH, como de tipo estructural con anomalías menores tipo dismorfismo
craneofacial y malformaciones en miembros superiores. Se realizó asesoramiento
genético a los padres y al caso Tabla 2. Valoraciones
y estudios realizados al paciente. índice por el alto riesgo de
repetición en la descendencia, el porcentaje de repetición no se describe con
exactitud en la literatura, sin embargo, este depende del tamaño y del material
citogenético involucrado en los segmentos cromosómicos reorganizados. Las
características fenotípicas de esta patología cromosómica y los hallazgos
encontrados en el paciente se resumen en la Tabla 3.
Discusión
Tal como se reporta en el caso, los hallazgos
dismorfofisiológicos del paciente son consistentes con los hallazgos
previamente reportados en anomalías cromosómicas estructurales no balanceadas.
En el caso de nuestro paciente se confirmó la cromosomopatía mediante estudio
citogenético, con posterior identificación de origen paterno.
Las manifestaciones fenotípicas comúnmente asociadas a la
pérdida de material genético del 5q22 se muestran en la Tabla 3; gran parte
de las características mencionadas anteriormente y que estuvieron presentes en
el caso índice permiten hacer una correlación entre su fenotipo y cuadro
clínico con otros casos reportados en la literatura mundial, cabe resaltar que
en el paciente estudiado se descartó la hipoacusia, las alteraciones de tipo
visual y las malformaciones congénitas cardiacas, renales y de vías urinarias.
Actualmente se establece que el cariotipo con bandeo Q y G
se debe realizar a todo paciente con retardo mental sin otras causas obvias,
múltiples dismorfias, patrón de crecimiento anormal como talla baja, fenotipos
clínicamente anormales, retardo en el neurodesarrollo, en sospecha clínica de
deleción, microdeleción o microduplicación cromosómica, enfermedades monogénicas
recesivas con herencia ligada al cromosoma X de una mujer, antecedentes
familiares de alteraciones cromosómicas estructurales entre otras;
adicionalmente se reporta que pacientes con retardo mental inexplicable,
retraso en el desarrollo psicomotor, autismo, malformaciones congénitas,
alteraciones cromosómicas balanceadas con cariotipo en pacientes con fenotipos
anormales y trastornos de impronta entre otros, se puede optar por la
hibridación genómica comparativa por microarreglos (aCGH) por sus siglas
en inglés (array Comparative Genomic Hybridization), esta técnica
detecta mayores alteraciones cromosómicas estructurales que el cariotipo en el
caso de déficit mental inexplicable (20-25% versus 9.5%), sin embargo, el
microarreglo no reemplaza el cariotipo cuando se tiene la sospecha de alguna
cromosomopatía ya que no permitiría distinguir entre un arreglo estructural y
una alteración de tipo numérica8,15; el cariotipo con Bandeo Q y G fue realizado al caso
índice por presentar un coeficiente intelectual muy bajo (CI: 52) según la
escala de inteligencia para niños Weschler, además de presentar
trastorno de la conducta tipo inatención e hiperactividad con características
dismórfofisiológicas claras, evidenciándose en el 100% de las metafases de las
células estudiadas una translocación no balanceada tipo t(5;7) (q22;p15) y
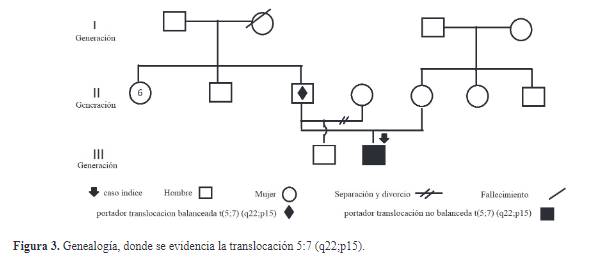
aCGH a los progenitores del La importancia de detectar portadores de
translocaciones balanceadas como es el caso del padre de nuestro caso índice,
es que el desequilibrio cromosómico puede llevar a las siguientes posibilidades
en futuros embarazos: 1) Heredarse únicamente los cromosomas normales, 2)
Heredarse la misma translocación paterna, 3) Heredar una translocación no
balanceada como lo fue en nuestro caso índice y por el cual se hace evidente
las manifestaciones clínicas o 4) Culminar en aborto9. Estas variantes se han
podido estudiar en gametos de pacientes con translocaciones recíprocas, en
donde se ha evidenciado que durante la profase-I de la meiosis, los cuatro
cromosomas que comparten segmentos homólogos de un portador de dicha
translocación adoptan una estructura referida como tetravalente; durante
la anafase-I esta estructura puede segregarse de tres formas: la primera es 2:2
que se subdivide en tres grupos de segregación a) alternante, b) adyacente-I
(en donde los centrómeros homólogos migran a polos opuestos de la célula) y c)
adyacente-II; la segunda es 3:1 y la tercera es 4:0 (en donde los centrómeros
homólogos migran a un mismo
En la segregación alternante se producen gametos cromosómicamente
balanceados con fetos portadores de la translocación (como lo es el caso del
padre de nuestro caso índice) con una frecuencia del 33 al 57%; en los otros
esquemas de segregación se generan gametos con cromosomas no balanceados de la
siguiente manera: en la segregación adyacente-I, el cromosoma translocado se
segrega con el cromosoma normal no homólogo involucrado en la translocación con
una frecuencia del 16 al 52%; para adyacente-II, el cromosoma translocado se
segrega con el cromosoma normal homólogo involucrado en la translocación con
una frecuencia del 0 al 39%; en los casos de la segregación 3:1 se genera un
gameto con 22 cromosomas y otro con 24 cromosomas con una frecuencia del 0 al
40%, y en la segregación 4:0 se genera un gameto con 21 cromosomas y otro con
25 cromosomas, esta última es considerada una alteración cromosómica
infrecuente17-19.
La frecuencia de descendencias con translocaciones no
balanceadas va a depender del tamaño y material citogenético involucrado en los
segmentos cromosómicos reorganizados, es por esto que muchos individuos
portadores de translocaciones balanceadas detectadas por descendencia portadora
de cambios cromosómicos no balanceados o por cualquier otra causa, acuden a
consultoría genética mostrando un interés particular en estipular el riesgo de
tener un linaje con anomalías congénitas y de fracaso reproductivo18,20, motivo por el cual
es de vital importancia informar, educar y dar un soporte a los individuos y
miembros de la familia que padecen de algún trastorno genético o que tienen
riesgo de padecerla.
Hay que tener en cuenta que cuando existen reordenamientos
en el material genético en especial si hay pérdida o ganancia de este, las
manifestaciones fenotípicas están ligadas a la localización de los puntos de
ruptura y como consecuencia a los genes que estén incluidos en estos segmentos.
En este caso, el paciente presenta punto de ruptura para originar la
translocación en el locus 5q21-q22 en el cual se localiza exactamente el gen
APC (Adenomatous Polyposis Coli)15; dicho gen tiene un tamaño de 8500 pb y
codifica una proteína de 2843 aminoácidos, los síndromes asociados a mutaciones
en la línea germinal de este gen incluyen la Poliposis Adenomatosa Familiar
(FAP por sus siglas en inglés) en donde se desarrollan más de 100 pólipos
adenomatosos con un 93% de riesgo de malignización a los 50 años, también FAP
atenuado (AAPC por sus siglas en inglés), consistente en un menor número de
pólipos o de presentación tardía, así como pueden desarrollar Síndrome de
Gardner y de Turcot que se diferencian de la FAP en que el primero además de
los pólipos presenta osteomas y tumoraciones de tejidos blandos y el segundo se
encuentra asociado al cáncer de colon y tumores del sistema nerviosos central
(generalmente meduloblastoma)21-23; por todo lo anterior, es importante realizar
estudios complementarios en el caso índice para descartar estas patologías.
Con respecto al cromosoma 7, su punto de ruptura y
translocación, en la literatura se describe: locus 7p15 donde se localiza el
gen DFNA5 (Deafness, Autosomal Dominant-5) cuya alteración puede llevar
a sordera autosómica dominante, en el locus 7p15-p13 se localizan los genes
CCM2 (Cerebral Cavernous Malformations-2), CGK (Glucokinase),
HOXA11 (Homeobox-A11) y HOXA13 (Homeobox-A13) las cuales pueden
llevar a malformaciones cavernosas cerebrales, hiperinsulinismo familiar,
sinostosis radio ulnar con trombocitopenia amegacariocitica y al síndrome
mano-pie-útero respectivamente, y finalmente en el locus 7p15-p14 se encuentra
el gen UMPH1 (Uridine 5´-Monophosphate Hydrolase-1) cuya alteración
puede inducir anemia hemolítica debido a la deficiencia de UMPH124; hasta el momento, en
la evolución clínica del paciente no se han encontrado hallazgos clínicos
relacionados con las patologías que describe la literatura respecto a la
reorganización genómica de este cromosoma.
Teniendo en cuenta todo lo anterior es importante que el
paciente permanezca en vigilancia médica constante debido al riesgo de
desarrollar las patologías descritas en la literatura para los cromosomas
implicados en la translocación y las consecuencias que dichas patologías puedan
tener en la salud y calidad de vida del paciente en un futuro, por todo lo
anterior se debe llevar un control semestral por psiquiatría y psicología, de
manera trimestral con terapia ocupacional, control por gastroenterología anual
por el riesgo de compromiso del gen APC y cada seis a doce meses con la
especialidad de endocrinología pediátrica.
Conclusión
La importancia de la identificación de los pacientes con
fenotipos sugerentes de alteraciones genéticas radica en la posibilidad de
brindar un manejo adecuado para contribuir a la mejora de la calidad de vida de
estos individuos, además de poder brindar consejería genética a la familia ante
la posibilidad de repetición de dichas alteraciones en nuevos miembros; para
esto, debe llevarse a cabo un estudio citogenético; actualmente se encuentran
disponibles diferentes pruebas diagnósticas como el cariotipo con bandeo Q y G,
el cual resulta muy útil en el estudio de individuos con dismorfismos,
trastornos del comportamiento, retardo mental o alteraciones en el
neurodesarrollo de origen desconocido, entre otras.
La localización de los sitios de ruptura e identificación de
los genes involucrados en los reordenamientos de material genético, así como la
pérdida o ganancia de éste es fundamental para establecer el riesgo potencial
del paciente para desarrollar patologías asociadas, motivo por el cual se hace
necesario la vigilancia médica de nuestro caso índice mediante la realización
de estudios complementarios como la prueba aCGH para determinar los
puntos de ruptura de la translocación y los genes involucrados en dicha
alteración.
Consideraciones éticas
Este reporte de caso recibió la autorización de los padres
del paciente estudiado.
Conflictos de interés
Los autores declaran no tener ningún conflicto de interés
Referencias
1.
Centeno Malfaz IF, Beltrán Pérez IA, Ruíz Labarga C, Centeno
Robles J, Macías pardal J, Martín Bermejo M. Cromosomopatías en recién nacidos.
An Esp Pediatr. 2001; 54(6): 582-587. doi: https://
doi.org/10.1016/S1695-4033(01)77598-4.
2.
Ospina-Ramírez JJ, Castro-David MI, Hoyos-Ortiz LK,
Montoya-Martínez JJ, Porras-Hurtado GL. Factores asociados a malformaciones
congénitas: En un centro de tercer nivel región centro occidental - Colombia
(ECLAMC). Rev Med Risaralda. 2018; 24(1): 14-22. doi: http://dx.doi.
org/10.22517/25395203.9317.
3.
Bueno ML. Cromosomas, vehículos en la organización y trnasmisión
de caracteres. Acta Biológica Colombiana. 2011;16(3): 43-60. http://
dx.doi.org/10.15446/abc.
4.
Oliver-Bonet M, Navarro J, Carrera M, Egozcue J, Benet J.
Aneuploid and unbalanced sperm in two translocation carriers: evaluation of the
genetic risk, Mol Human Reprod. 2002; 8(10): 958-963. doi:
https://doi.org/10.1093/molehr/8.10.958.
5.
Monjagata N, Ascurra M, Herreros MB, de Torres E. Retardo mental
y cardiopatía en una portadora de una translocación balanceada. Mem Inst Inv
Cienc Salud IICS. 2005; 3(1): 58-60.
6.
Caballín MR, Miro R, Egozcue J. Abnormal phenotype in a child
with the same balanced translocation (5;7)(p15;q22) as his father. Clin
Genetics. 1981; 20(6): 428-431. doi: https://doi.
org/10.1111/j.1399-0004.1981.tb01053.x.
7.
Tharapel A, Summit R, Wjlroy R, Martens P. Apparently balanced de
novo translocations in patients with abnormal phenotypes: report of 6 cases.
Clin Genet. 1977; 11(4): 255-269. doi: https://
doi.org/10.1111/j.1399-0004.1977.tb01310.x.
8.
Esparza-Garcia E, Cardenas-Conejo A, HuicocheaMontiel J,
Araújo-Solís M. Cromosomas, cromosomopatías y su diagnóstico. Rev Mex
Pediatr. 2017; 84(1): 30-39.
9.
Espinosa-Álvarez D, Guerrero-Jordan D, FernándezCastillo O.
Aberración cromosómica balanceada. A propósito de un caso. Mul Med. 2013;
17(4): 1-8.
10.
Tamar Silva C, Contreras NC, Fonseca DJ. Utilidad de la
citogenética en la medicina actual. Visión histórica y aplicación. Acta Médica
Col. 2008; 33(4): 309-316.
11.
Artigas-Pallarés J. Tratamiento farmacológico del retraso mental.
Rev Neurol. 2006; 42 (Supl 1): S109-S115. doi: https://doi.org/10.33588/
rn.42S01.2005702.
12.
Portela Sabari A, Carbonell Naranjo M, Hechavarría Torres M,
Jacas García C. Trastorno por déficit de atención e hiperactividad: algunas
consideraciones sobre su etiopatogenia y tratamiento. MEDISAN. 2016; 20(4):
553-563.
13.
González Meneses A. Dismorfología clínica y genética I: enfoque
diagnóstico del paciente dismórfico. An Pediatr Contin. 2008; 6(3): 140-46.
doi: https://doi.org/10.1016/S16962818(08)74868-6.
14.
Understanding rare chromosome disorders: Síndrome relacionado al
gen PACS1. Unique; 2018. https://www.rarechromo.org.
15.
Heald B, Moran R, Milas M, Burke C, Eng C. Familial adenomatous
polyposis in a patient with unexplained mental retardation. Nat Clin Pract
Neurol. 2007; 3(12) 694-700. doi: https://doi. org/10.1038/ncpneuro0658.
16.
Godo A, Vidal F, Blanco J, Anton E. Desequilibrios cromosómicos
en espermatozoides de individuos portadores de translocaciones recíprocas. Rev
Asoc Est Biol. 2012; 17(1):12-16.
17.
Morel F, Douet-Guilbert N, Le bris MJ, Herry A, Amice V, Amice J,
et al. Meiotic segregation of translocations during male gametogenesis. Int J
Androl. 2004; 27(4): 200-212. doi: https://doi.
org/10.1111/j.1365-2605.2004.00490.x.
18.
Aguilar J, Sainz L, Mitjans M. Estimado teórico de riesgo de
tener descendencia no balanceada, en portadores de translocaciones recíprocas.
Centro Nacional de Genética Médica. 2008. Disponible en línea en:
https://bit.ly/2Oxl7uf
19.
Wiland E, Midro AT, Panasiuk B, Kurpisz M. The analysis of
meiotic segregation patterns and aneuploidy in the spermatozoa of father and
son with translocation t(4;5)(p15.1;p12) and the prediction of the individual
probability rate for unbalanced progeny at birth. J Androl. 2007; 28(2):
262-272.
doi: https://doi.org/10.2164/jandrol.106.000919.
20. Midro
A, Wiland E, Panasiuk B, Lesniewicz R,
Kurpisz M. Risk Evaluation of Carriers With
Chromosome Reciprocal Translocation t(7;13) (q34;q13) and
concomitant meiotic segregation analyzed by FISH on Ejaculated Spermatozoa. Am
J Med Gene A. 2006; 140: 245-256. doi: https://doi. org/10.1002/ajmg.a.31083.
21.
Scott R, Froggatt N, Trembath R, Evans D, Hodgson S, Maher E.
Familial infiltrative fibromatosis (desmoid tumours) (MIM135290) caused by a
recurrent 3´ APC gene mutation. Hum Mol Genet. 1996; 5(12): 1921-1924. doi:
https://doi. org/10.1093/hmg/5.12.1921.
22.
Passalacqua C, Aravena T, Castillo S. Genética del cáncer de
cólon. Rev Hosp Clin Univ Chile. 2010; 21: 162-169. https://bit.ly/2R5reHF.
23.
Yamaguchi T, Koizumi K, Arai M, Tamura K, Lijima T, Horiguchi S,
et al. A large deletion of chromosome 5q22.1-22.2 associated with sparse type
of familial adenomatous polyposis: report of a case. Jpn J Clin Oncol. 2014;
44(12): 1243-1247. doi: https://doi.org/10.1093/jjco/hyu150.
24.
Gilbert F. Disease Genes and chromosomes: disease maps of the
human genome. chromosome 7. Genet Test. 2002; 6(2): 141-6.
https://doi.org/10.1089/ gte.1999.3.243